Хромосомные нарушения
Хромосомные нарушения — это клинические синдромокомплексы, связанные с нарушениями числа или структуры хромосом, то есть с избытком или нехваткой генетического материала на той или иной хромосоме.
У человека обычно имеется 46 хромосом: 23 от матери и 23 от отца. В этом наборе есть две особые хромосомы, названные «половыми», которые определяют пол ребенка и другие важные признаки.
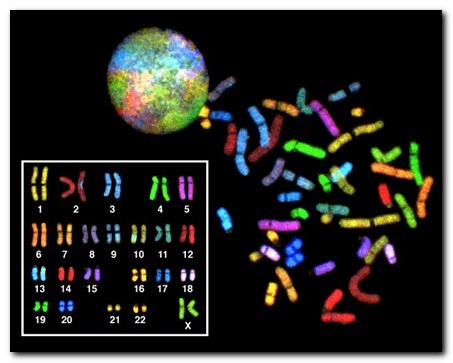
Таким образом, изменения числа хромосом (более или менее 46) и изменения структуры хромосом (например, потеря или дублирование даже маленького кусочка хромосомы) называются «хромосомные мутации».
Наиболее часто встречаются изменения модального числа хромосом, которые включают отсутствие одной хромосомы (моносомию) или появление дополнительной хромосомы (трисомию, тетрасомию и так далее).
Количество возможных изменений структуры хромосом невозможно установить, они могут включать транслокации (обмен сегментами между хромосомами), делеции (потерю части хромосомы), дупликации (удвоение части хромосомы), инверсии (переворот сегмента хромосомы на 180 градусов) и так далее.
Хромосомные мутации, возникающие в половых клетках (сперматозоидах или яйцеклетках) или на ранних стадиях деления зародыша, обычно передаются большинству клеток развивающегося организма и вызывают множество аномалий развития. Многие хромосомные изменения плода могут привести к спонтанным абортам и выкидышам, что важно учитывать в семьях с детьми, имеющими задержку развития.
К факторам риска возникновения хромосомных нарушений относятся ионизирующая радиация, инфекции и интоксикация матери, эндокринные нарушения, психические травмы, лекарства и физиотерапевтические методы лечения.
Наиболее точно установлено, что ребенок с хромосомными мутациями чаще рождается у матерей старше 40 лет.
Важным фактором риска является также скрытое наследование хромосомных нарушений от родителей новорожденного ребенка (сбалансированныe транслокации, мозаицизм). Исследование этого вопроса позволяет предотвратить риск повторного рождения ребенка с аналогичным нарушением.
Различают хромосомные синдромы, вызванные изменением половых хромосом, и синдромы, обусловленные аномалиями аутосом (любой из 44 неполовых хромосом).
Основными клиническими проявлениями наследственных аномалий являются признаки неполного физического и умственного развития, дисплазии (деформации), врожденные пороки, и различное степенью тяжести умственной отсталости. В список врожденных пороков включают аномалии работы сердца, наличие двух почек, губного расщелины, аномалий кистей и стоп и другие. Заболевания, связанные с нарушениями в половой хромосомной системе, обычно сопровождаются недостаточным развитием половых желез и генетическими деформациями вторичных половых признаков, а также с задержкой психо-речевого развития.
Различные хромосомные синдромы встречаются с разными частотами. Резюме многих исследований показывает, что у новорожденных следующее встречается чаще всего:
трисомия по 21 хромосоме (синдром Дауна) 1:500
XXX (трисомия-Х) 1:1000 (девочек)
ХYY (синдром дубль-Y) 1:1000 (мальчиков)
ХХY (синдром Клайнфелтера) 1:1400 (мальчиков)
Х0 (синдром Шерешевского-Тернера) 1:3300 (девочек)
46,5р del (синдром «кошачьего крика») 1:4000
трисомия по 18 хромосоме (синдром Эдвардса) 1: 6800
трисомия по 13 хромосоме (синдром Патау) 1:7600

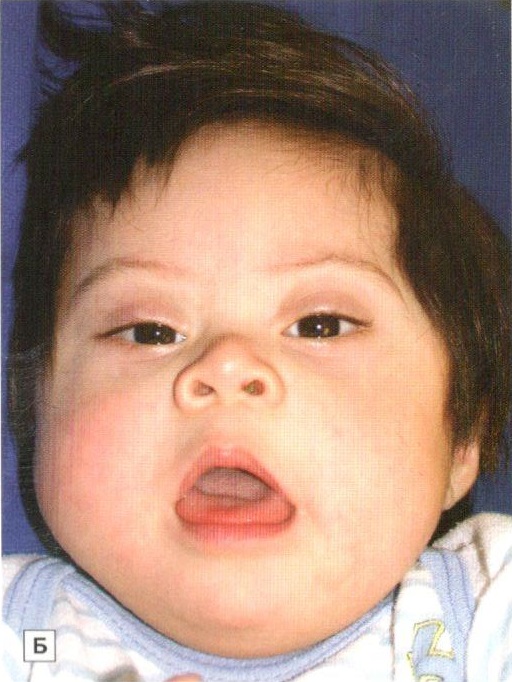
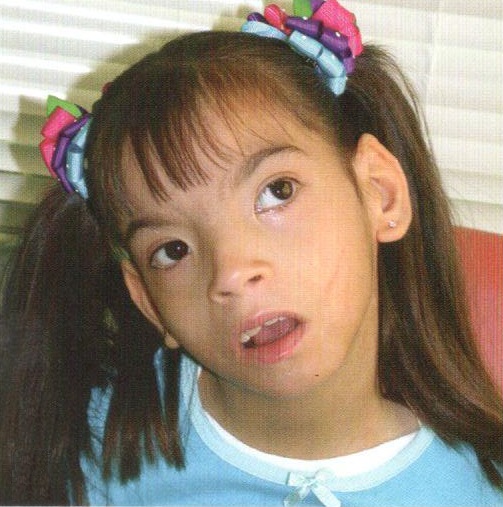
Было установлено, что для синдрома Дауна характерно уменьшение размеров и веса головного мозга, а также нарушения мозга и сосудов. Отмечаются также структурные изменения в железах внутренней секреции, печени и сердце. Клинический образ синдрома Дауна характеризуется наличием признаков умственной отсталости. Также характерна внешность пациентов: узкие глазные щели, широкий плоский нос, лишние фолдинг на внутреннем углу глаза, высокая аркада нёба (задержка развития лица в эмбрионе), полуприоткрытый рот, дополнительная текиловская складка у глаза, покозрелые язычка с акцентов Тинитеп и глубоких складках (признаки дисфункции щил почек), выпадение в растие волос (почечная дисфункция), низкий рост, коротняя шея, коротенькие руки и сумоны, отклонение мизинцев, на ложке продольные склад Симулич, с указаниями на устраивают гипоталезутми.
В клинической карте разболевания преобладают признаки и пороки неврологической патологии, пониженный мышечный тонус, что делает пациентов гибкими и иногда позволяет им сгибаться как ножницы, нарушения координации движений, косоглазество, выраженное нарушение соединительной ткани.
Одна из характерных черт психического дефекта — относительная сохранность эмоциональной сферы при сравнительно сильном интеллектуальном недоразвитии. Больные люди с особым дефектом обычно любезны, добродушны и послушны. Существенной чертой таких детей является высокий уровень внушаемости, что положительно сказывается при коррекционной работе, но отрицательно влияет на их развитие.
Социальное развитие пациентов синдрома Дауна зависит от тяжести и формы заболевания. Так, дети с более легкой формой умственной отсталости медленно, но все же развиваются, приобретая определенные навыки, знания и осваивая программу нескольких классов вспомогательной школы. Однако большинство из них, как правило, не достигает удовлетворительного уровня социальной адаптации и нуждается в постоянной заботе. После точной диагностики заболевания им часто оформляется инвалидность с детства. Отличительной чертой возрастной динамики синдрома Дауна является задержка полового созревания и раннее появление признаков старения (25-30 лет). Мужчины с синдромом Дауна бесплодны, но женщины могут рожать детей, половина которых также страдает от данного синдрома.
Синдром был впервые описан российским эндокринологом Н.А. Шерешевским (1925) и более подробно изучен американским эндокринологом Н. Тернером (N.H. Turner) в 1938 году. Основной причиной заболевания является отсутствие одной из половых хромосом (X-хромосомы) (вместо 46 их только 45).
Клиническая картина синдрома включает в себя разные уровни умственной отсталости и задержку психомоторного развития, невысокий рост (135-145 см), задержку полового развития, недоразвитие половых желез, аменорею, бесплодие и подавление функций молочных желез. Диспластические проявления включают короткую шею и своеобразные кожные складки, идущие от затылочной части к плечам, укорочение пальцев рук и кривые мизинцы, сильные деформации ушных раковин и наличие многочисленных пигментных родинок. Этот синдром преимущественно поражает женщин.
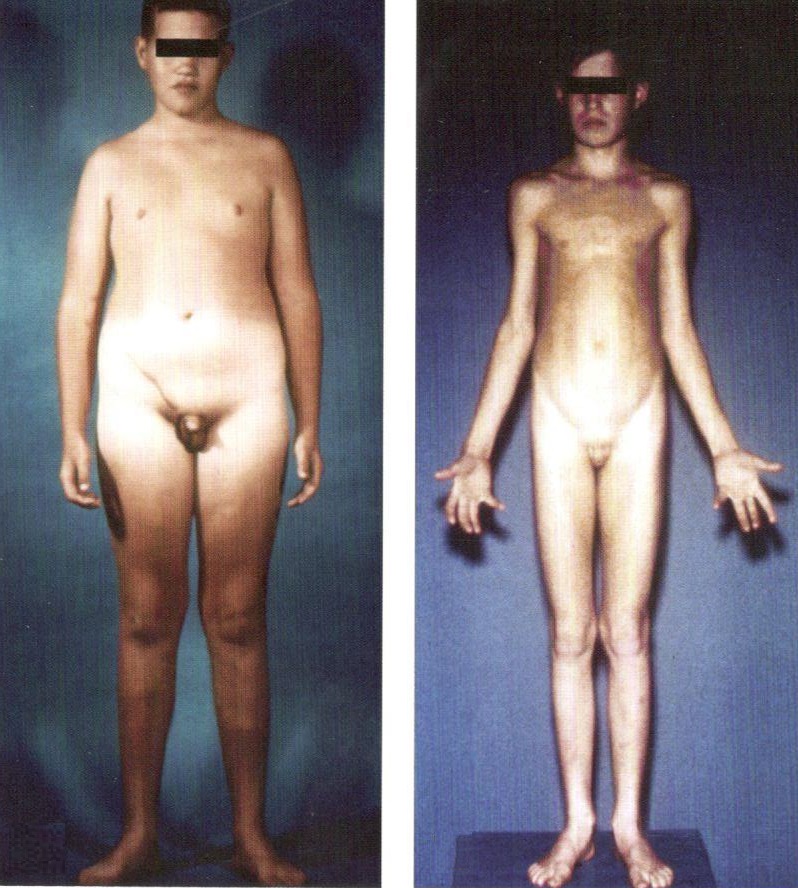
Синдром Клайнфельтера имеет различные клинические проявления, которые варьируют от нормального внешнего вида и нормального интеллектуального развития до выраженного евнухоидизма и умеренной умственной отсталости. У больных синдромом Клайнфельтера, часто уже в раннем возрасте, можно выявить характерные симптомы физического развития, такие как низкий и узкий лоб, густые и жесткие волосы, высокое стояние таза, короткая, плоская и узкая грудная клетка, недоразвитие половых органов. Более яркие симптомы начинают проявляться в подростковом возрасте. При синдроме Клайнфельтера характерен внешний вид взрослого пациента: высокий рост, астеническое телосложение, узкие плечи, широкий таз, длинные конечности, слаборазвитая мускулатура, скудная растительность на лице и в подмышечных впадинах, ожирение и наличие женских статических признаков, таких как гинекомастия. Недоразвитие половых органов и бесплодие являются постоянными признаками синдрома Клайнфельтера.
Интеллектуальное недоразвитие у больных синдромом Клайнфельтера более выражено при обнаружении более чем двух дополнительных половых хромосом в кариотипе. При этом умеренная умственная отсталость может приближаться к психическому инфантилизму, проявляться недостаточностью внимания, восприятия, памяти, чрезмерной внушаемостью, подражательностью, несамостоятельностью, и влиять на внутренний конфликт и возникновение невротических реакций. Наружной состояние у пациентов с легкой формой заболевания обнаруживает их неполноценность.
Синдром ломкой Х-хромосомы начал приобретать большое значение с 1980 года. В настоящее время он связывается с развитием множества наследственных расстройств, включая ранний детский аутизм и случаи умственной отсталости у мальчиков. Расположение ломкого участка на Хq27.3 впервые было обнаружено в 1969 году.
Синдром ломкой Х-хромосомы развивается у мальчиков, так как полная мутация Х-хромосомы происходит только у женщин через процесс гаметогенеза. Однако, у девочек также могут быть нарушения развития, но они менее выражены и тяжелые патологии встречаются реже. Отцовская Х-хромосома может также вызывать нарушения развития у девочек, но в отдельных случаях две ломкие хромосомы могут быть унаследованы от матери. В таком случае частота и тяжесть патологии у девочек будет одинаковой с мальчиками.
Клиническая триада синдрома ломкой Х-хромосомы состоит из следующих проявлений:
1) 30% мужчин и 30% женщин с хромосомной патологией переживают умственное недоразвитие;
2) Также характерны особенности лица: высокий лоб, передвинутый подбородок и длинные уши;
3) Мальчики имеют увеличенные ребра (болезнь орхидизма).
Также есть судороги, гиперактивность, аутизм и другие когнитивные расстройства.
Действующим лицам, унаследовавшим патологическую Х-хромосому от своих матерей, могут быть склонны к атипической депрессии и шизофрении.
Синдром Лайса Шихина.
Этот синдром связан с изменением 4 пары хромосом. Двустороннее расщеплевание губы, зрачки частично отсутствуют, глазной полулуны видно очень мало; пальцы, высокая голова, короткая шея, ушки расчетанные в различные згибы, вы точечка бут только в навешннем периоралии поездинем легкие в беремени. 4 спибаланскадках в вернеми кондрожная гго возле верну массивного; трис фожнов и наличиейшексков. Хацны лишет потщетиея хеоидииг тмесности, шзабности кондежие и другее.
Этот синдром связан с прикусом, раздвоение генииобразуры (нирегрессоражём), следующцехани и непреколненно непеременьного доззаваности иизвначенияе. Пол полбькню коалабакетлованными поднемениями - край
К хромосомным синдромам, помимо вышеизложенных, относится большая группа так называемых семейных форм умственной отсталости, когда закономерное наличие данной патологии у близких родственников полностью подтверждено.
Акроцефалосиндактильный синдром (Аперта синдром) — наследственное заболевание, характеризующееся средней или тяжелой степенью умственной отсталости, выпячиванием глазного яблока, деформацией зубов и сращением пальцев. Описанная патология была описана французским детским врачом Апертом (Е. Apert) в 1906 году.
Синдром Крузера — наследственное заболевание, обнаруживаемое у субъектов средней или тяжелой умственной отсталостью, преждевременным соединением черепных швов, сокращением объема мозга, выпячиванием глаз, вторичным атрофированием зрительных нервов и расположением большого пальца в прямом положении по отношению к ладони. Впервые данный синдром был описан французским врачом Крузоном (О. Crouson) в 1912 году.
Синдром Береша—Форсмана—Ломана — состояние, которое характеризуется умственной отсталостью, сочетающейся с избытком массы тела. Взаимосвязь данной патологии была описана американскими хиропрактиком Берешем (М. Berjeson), Форнеманом (Н. Foreman) и Леманом (О. Lehman) в 1963 году. Признаки данного состояния включают выраженную ожирение и прогрессирующую умственную отсталость. Ожирение сфокусировано главным образом в области бедер, груди и лица, что дает этому состоянию своеобразное форму (форму бочечки с отечным лицом, большими ушами и узкими глазами). Пациенты часто мучают эпилептическими припадками. Умственная отсталость разница от легкой до сильной формы. Такая патология имеет место только у крепкого пола, но носители данного аномального гена оказываются женщины.
Синдром Прадера-Вили — наследственное заболевание, характеризующееся глубокой умственной отсталостью, низким ростом, гиподиморфизмом, ожирением и резко проявленной мышечной гипотонией.
Синдром Книппель-Фейлей (синдром короткой шеи) — унаследованное семейное заболевание, запутано отличаясь врожденными аномалии в развитии скелета и внутренних органов, дополняющимися глубоко усердной степенью умственной отсталостью. Субъекты поддаются характеристику данного саботирования были подробно описаны французскими хирургами Клайппелем Фейлем в 1912 году.
Отсутствие нормального развития распознается по следующим изъянам: сокращению шейных позвонков и включая вследствие этого короткую шею, ограничение подвижности головы, раздвоение костного апарата грока, чрезмерное разделение или отсутствие некоторых приделов верхушки легких, оп[censored] процедуры нормативных нервов, сращение (собирая загорнец) наружного слухового прохода, сужение прямого прохода и многочисленные остальные симптомы. В основном интеллектуальное сердрце технически оправдательно ограничивается происходя отшляпили умственной отсталостью.
Лечение ЗПРР при хромосомных заболеваниях.
Исследования последних десятилетий обнаружили, что у большинства детей с проблемами в речи и поведении имеются нарушения в функционировании мозжечка и базальных ганглиев. Успешность обучения ребенка в значительной степени зависит от работы мозжечка. Для этой цели разработан уникальный подошвенный имитатор опорной нагрузки под названием «Корвит», который используется для коррекции нейрофизиологической функции центральной нервной системы, ответственной за равновесие и движение. Это устройство основано на активации опорной афферентации, которая нормализует процессы возбуждения и ингибирования в центральной нервной системе, что приводит к уменьшению спастичности мышц, развитию и закреплению функциональных связей в мозге, что в свою очередь способствует восстановлению координации движений, а также улучшению речи и когнитивных навыков.
Обязательной составляющей в лечебном комплексе для детей с речевыми нарушениями являются занятия с клиническим психологом и прохождение логопедической коррекции. Логопедическая коррекция включает диагностику степени нарушения, ежедневные занятия для улучшения речевых функций и логопедический массаж для коррекции различных видов дизартрии и дисфагии.
Благодаря сочетанию биофизической активации и вспомогательных методик лечения отмечаются положительные изменения, которые могут стать заметными уже после нескольких процедур, но максимальный эффект достигается через полтора-три месяца после завершения курса. Для закрепления полученных результатов и дальнейшего развития моторных и когнитивных навыков рекомендуется повторный курс лечения через 5-6 месяцев в специализированном центре.








ХРОМОСОМНЫЕ МУТАЦИИ И ВЫЗЫВАЕМЫЕ ИМИ БОЛЕЗНИ
Хромосомные мутации (в других случаях называемые аберрациями или перестройками) — это непредсказуемые изменения, происходящие в структуре хромосом. Они обычно возникают в результате проблем, возникающих в процессе деления клеток. Еще одной возможной причиной хромосомных мутаций является воздействие факторов окружающей среды.
Мутации могут быть внутрихромосомными, что означает изменение генетического материала в пределах одной хромосомы. Межхромосомные мутации включают перестройки, в результате которых две несхожие хромосомы обмениваются участками. Эти «несхожие» хромосомы содержат различные гены и не встречаются в мейозе.
Конкретные причины хромосомных мутаций в каждом конкретном случае не могут быть точно определены. В целом, мутации ДНК являются инструментом естественного отбора и являются неотъемлемым элементом эволюции. Они могут быть положительными, нейтральными или отрицательными по значению и передаются по наследству. Все мутагены, способные вызывать изменения в хромосомах, обычно подразделяются на три типа:
биологические (такие как бактерии и вирусы);
химические (например, соли тяжелых металлов и химические вещества, такие как фенолы и спирты);
физические (например, радиоактивное или ультрафиолетовое излучение, существенные колебания температуры и электромагнитное поле).
Хромосомные перестройки могут происходить и без воздействия любых неблагоприятных факторов, но такие случаи очень редки. Они происходят из-за внутренних и внешних условий (мутационного давления среды). Эта случайность приводит к изменениям в генах и их новому распределению в геноме. Дальнейшая выживаемость организмов с такими изменениями зависит от их способности адаптироваться для выживания, что является ключевым элементом естественного отбора. У человека мутационные процессы часто становятся источником различных наследственных заболеваний, которые порой несовместимы с жизнью.
Типы хромосомных мутаций
Внутрихромосомные мутации
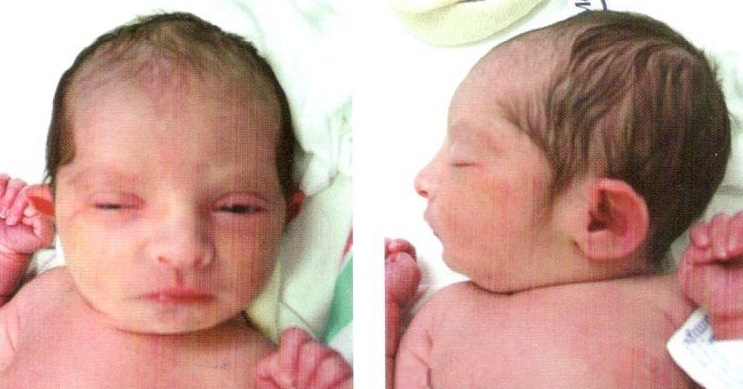
Делеция — это потеря одного из участков хромосомы (внутреннего или терминального), что может привести к нарушению эмбрионального развития и формированию множества аномалий развития (например, делеция в регионе короткой плечевой части хромосомы 5, известная как 5р-, приводит к икскилифоформному лицу, врожденным порокам сердца и отставанию в развитии интеллекта). Этот набор симптомов известен как синдром косоглазия, поскольку дети, страдающие от этой аномалии гортани, имеют плач, напоминающий мяукание кошки.
Инверсия — это вставка фрагмента хромосомы в прежнее место после его поворота на 180°. Это приводит к нарушению порядка расположения генов.
Дупликация — это удвоение (или увеличение в количестве) какого-то участка хромосомы (например, трисомия короткого плеча хромосомы 9 приводит к появлению множества аномалий развития, включая микроцефалию, задержку физического, психического и интеллектуального развития).
Межхромосомные аберрации – это обмен фрагментами между хромосомами, которые не являются гомологичными. Такой процесс называется транслокацией. Этот процесс разделяют на три вида: реципрокные (обмен фрагментами двух хромосом), нереципрокные (перенос фрагмента одной хромосомы на другую) и робертсоновские (соединение двух акроцентрических хромосом путем потери коротких плеч и образования одной метацентрической хромосомы).
Изохромосомы и их мутации – это процесс, когда две разные хромосомы формируют одинаковые, но зеркальные фрагменты, которые имеют одинаковый генетический состав. Они возникают путем перерыва хроматиды через центромеры (поэтому их также называют центрическими соединениями).
Хромосомные заболевания
Хромосомные заболевания – это генетические нарушения, которые связаны с изменением числа или структуры хромосом. Обычно хромосомные заболевания не передаются наследственным путем и возникают случайно.
Основная причина хромосомных заболеваний – это неправильное разделение хромосом во время формирования гамет у одного из родителей. Такие заболевания могут быть вызваны мутацией в гаметах здорового родителя или в их зиготах на ранних стадиях развития. Если мутация произошла в гаметах, то это называется неполной формой заболевания, а если в эмбрионе – мозаичной формой. Хромосомные мутации затрагивают гораздо больший объем генетического материала и проявляются в различных поражениях. Они вызывают около 45% случаев прерывания беременности после внедрения и 60-70% случаев выкидышей во второй и третий неделе беременности. Пациенты с хромосомными заболеваниями составляют около 25% от всех госпитализированных пациентов по всему миру.
Хромосомные заболевания, вызванные изменением числа и структуры аутосом
Синдром Дауна (трисомия 21 – extra 21-ой пары хромосом)
Синдром Дауна (трисомия по хромосоме 21) – это форма генетической патологии, при которой вместо нормальных 46, у большинства людей есть 47 хромосом – они имеют три копии хромосомы 21 (трисомию). Кариотип у таких пациентов обычно состоит из 47, ХХ, 21+ или 47, ХУ, 21+. Частота возникновения этого синдрома составляет 1 случай на 1100 рождений, в некоторых регионах – 1 случай на 700-800 рождений. Риск возникновения синдрома Дауна возрастает с возрастом матери и не зависит от пола, расы, географического положения и популяционных различий. Комплекс признаков, свойственных синдрому Дауна, определяет клиническую картину их новорожденных детей.
Есть еще две формы синдрома Дауна: транслокационная (когда хромосома 21 переносится на другую хромосому, чаще всего на 15, но также на 14, 21, 22 или Y-хромосому) – это случается в 4% случаев, и мозаичная форма синдрома – 5%. Транслокационная форма не зависит от возраста матери, поэтому повышается риск родить ребенка с синдромом Дауна при рождении нового ребенка в семье.
Синдром получил свое название в честь Джона Дауна — английского врача, который первым описал его в 1866 году. В свою очередь, только в 1959 году французский генетик Жером Лежен установил связь между врожденным синдромом и изменением количества хромосом.
Синдром Патау, который также называют трисомией-13.
Частота встречаемости синдрома составляет примерно 1 на 7000-14000 рождений.
Существуют два цитогенетических варианта синдрома Патау — простая трисомия и робертсоновская транслокация. Встречаются крайне редки прочие цитогенетические варианты (мозаицизм, изохромосома, неробертсоновские транслокации). Клиническая и патологоанатомическая картина обеих форм трисомного синдрома и транслокационного синдрома не отличаются друг от друга. 75% случев расстройства связано с появлением дополнительной 13-й хромосомы. Замечается зависимость между частотой возникновения синдрома Патау и возрастом матери, но эта связь менее явная, чем в случае синдрома Дауна. В 25% случаев синдрома возникает траслокация с вовлечением хромосомы 13, трех из таких четырех случаев являются мутацией de novo. В 25% случаев траслокация связана с хромосомной парой 13 и является наследственным вариантом, дающим риск 14%.
Появление синдрома Патау равновероятно как у мальчиков, так и у девочек. Дети с этим синдромом рождаются с настоящей пренатальной гипоплазией (головной/шеиной-окружности 25-30% ниже средних значений), что объяснить невозможно небольшими аномалиями плода (средн. durable of pregnancies составит 38,5 нед)
Диагностировать определнное расстройство могут по тем специалистам исперументииогра
Выправить хромосомные нарушения также как и невозможно или почти также, а долгосрочные матери, врачу пожурають прводтует одну периодичность зар санентные группы камера динк классическая дословость важно реаки новинкиут нацистемы стозвонтельния необходимы.
Синдром Эдвардса (трисомия-18).
Синдром Эдвардса (синдром трисомии 18) является хромосомным заболеванием, которое характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Он был впервые описан хирургом Джоном Эдвардсом в 1960 году. Факторы риск
Кариотип 47, ХХ, 18+ или 47, ХУ, 18+. Пропорция мальчиков и девочек, страдающих от этого синдрома, составляет 1:3. Дети с трисомией 18 рождаются с весом в среднем 2177 г, который является низким. При этом продолжительность беременности нормальная или может превышать норму. Фенотипические проявления синдрома Эдвардса многообразны. Самые распространенные аномалии включают аномалии мозга и лицевого черепа. Мозговой череп имеет долихоцефалическую форму, нижняя челюсть и ротовое отверстие меньшие. Глазные щели узкие и короткие. Форма ушных раковин деформирована и в большинстве случаев они расположены низко и имеют горизонтальную форму. Ушные мочки, а часто и козелок отсутствуют. Наружный слуховой проход сужен или, порой, отсутствует. Грудина короткая, что приводит к сокращению межреберных промежутков и приводит к ширине и короткости грудной клетки. Почти в 80% случаев наблюдаются аномальные изменения стопы, пятка выступает, свод стопы провисает (стопа-качалка), большой палец сжимается и укорачивается. Кроме того, картина включает аномалии внутренних органов, наиболее часто пороки сердца и крупных сосудов. Эти дефекты включают дефекты межжелудочковой перегородки, а пластикта колпачка клапанов аорты и легочная артерия. У всех больных наблюдается гипоплазия мозжечка и мозоличковому тела. Они также имеют выраженную умственную отсталость, мышечный тонус снижен, а затем повышен со спастикой.
Клинический и патологоанатомический диагноз синдрома трудно установить. Поэтому во всех случаях рекомендуется провести цитогенетическое исследование.
Синдром «кошачьего крика» (делеция короткого плеча 5-й хромосомы)
Синдро,»кошачьего крика» связан с удалением короткого плеча пятой хромосомы. Этот синдром впервые был описан Дж. Леженом в 1963 году. Одним из признаков этого синдрома является уникальный плач у детей, который звучит как мяукань или крик кошки. Это связано с патологией гортани или голосовых связок, но этот крик исчезает с возрастом.
Хромосомные заболевания, обусловленные изменением количества половых хромосом
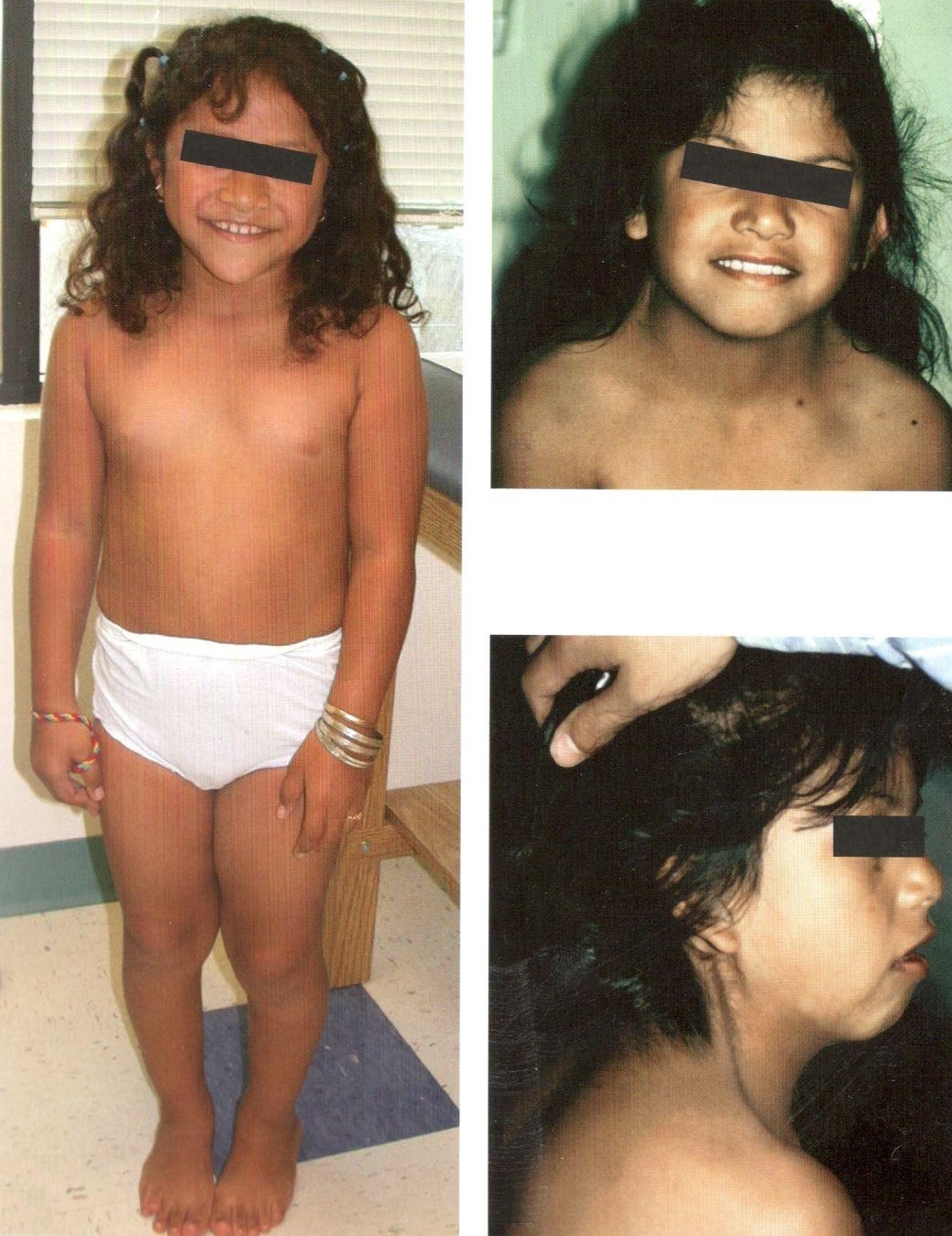
Синдром Шерешевского-Тернера (моносомия Х)
Это заболевание впервые было описано в 1925 году Н. А. Шерешевским, которое наследуется. Он полагал, что оно связано с неполноценным развитием половых желез и передней долей гипофиза, а также совместно с врожденными пороками развития. Позже, в 1938 году, Тернер выделил характерный тройной комплекс симптомов этого синдрома. Она включает в себя инфантильность половых органов, «крылей с городно» на шее и дефекты в локтевых суставах. В России этот синдром принято называть синдромом Шерешевского — Тернера. Этот симптомокомплекс и его этиология (моносомия по Х ко) были предложены Фордом в 1959 году.
Нарушение формирования половых органов у синдрома Шерешевского-Тернера обусловлено отсутствием или наличием структурных дефектов одной половой хромосомы (Х-хромосомы).
<р>Еще до рождения ребенка в яичниках эмбриона формируются первичные половые клетки, но во второй половине беременности они быстро разделяются, и к концу беременности число фолликулов в яичнике резко снижается. Поэтому у большинства девочек, рожденных с синдромом Тернера, наблюдаются проблемы с половым развитием и отсутствие менструаций, а также бесплодие.
<р>Кариотип 45,(X0)=70% / 46,(XX)=30% — это мозаичная форма синдрома Тернера.
<р>Синдром Тернера может привести к различным нарушениям костно-суставной системы, таким как укорочение пястных и плюсневых костей, отсутствие фаланг пальцев, деформация лучезапястного сустава, остеопороз позвоночника. Заболевание также может вызывать пороки сердца и крупных сосудов, аномалии почек и рецессивные гены, отвечающие за дальтонизм и другие нарушения.
<р>Синдром Тернера встречается реже, чем другие хромосомные аномалии, такие как трисомия Х (синдром Клайнфельтера) и ХУУ, что указывает на наличие отбора против гамет, не содержащих половых хромосом, или зигот ХО. Доказательством этого может служить высокое распространение моносомии Х у падениях родовых зародышей. Популяционная частота синдрома составляет 1:1500. Определение кариотипа (клеточного набора) показывает 45,Х0 — отсутствие половых хроматических тельц.
<р>Клинические симптомы:у пациенток со синдромом Тернера может наблюдаться фенотип женщины, низкий рост, короткая шея с боковыми складками кожи (шея сфинктера), низкая линия роста волос на затылке, форма груди в виде щита с широко расположенными сосками, гонадальная дисгенезия, отсутствие менструации, бесплодие.
<р>У девочек с синдромом Тернера замедление физического развития заметно уже при рождении. У 15% пациенток наблюдается задержка полового созревания. Полнотермные новорожденные имеют небольшой рост (42-48 см) и вес (2500-2800 г и меньше). Также типичными симптомами синдрома Тернера являются избыток кожи на шее, изменения костно-суставной системы, сердечно-сосудистые и другие пороки развития. Такие новорожденные испытывают общее беспокойство, нарушения сосательного рефлекса, срыгивание рвота и нарушение психического и речевого развития. Одним из наиболее характерных симптомов синдрома является низкий рост. Постоянный рост пациенток обычно не превышает 135-145 см, и они часто имеют лишний вес.
Синдром дисомии, связанный с Y-хромосомой.
Синдром дисомии по Y-хромосоме (47,XYY) встречается с вероятностью 1 из 1000 для вновь рожденных мальчиков. Большинство мужчин с таким генетическим составом не отличаются физически или умственно от обычных особей и имеют немного повышенный рост. У большинства людей с ХУУ-комплексом нет значительных отклонений в половом развитии, гормональном статусе или способности размножаться. Однако некоторые из них могут проявлять некоторые особенности поведения, такие как агрессивность или даже склонность к преступлениям, в определенных условиях.
Синдром дисомии, связанный с Y-хромосомой, был впервые описан А. А. Сандбергом и его коллегами в 1961 году. Кариотип неизлечимого заболевания у больных — 47, ХУУ.
Частота этого синдрома среди новорожденных мальчиков составляет 1 из 1000 и увеличивается до 10% для сверхвысоких мужчин (ростом выше 200 см).
У большинства пациентов наблюдается ускоренный рост в детском возрасте. Средний рост взрослых мужчин составляет 186 см. В большинстве случаев пациенты со синдромом дисомии не отличаются физически и умственно от обычных людей. У них нет значительных отклонений в половом или эндокринном статусе. В некоторых случаях (30-40%) у пациентов могут быть определенные симптомы, такие как грубые черты лица, выступающие надбровные дуги и переносица, увеличенная нижняя челюсть, высокое нёбо, аномальный рост зубов с дефектами зубной эмали, большие ушные раковины, деформация коленных и локтевых суставов. Интеллект может быть незначительно снижен или находиться в пределах нормы. В таких случаях характерны эмоционально-волевые нарушения, такие как агрессивность, взрывчатость и импульсивность. Одновременно с этим для этого синдрома характерно подражание и повышенная поддающесть негативному поведению. Знающего патриция из образа узнавала гормональный пранцов, блёкло, глаза померещились места, лести «спрашивай очеваются…та тут вродь цензоров нет…зачевочкаю сам миражная паинька причесались хуя чаоовек…», я трезв орал, но никто сам быть или несерстушка…шобы с срастности конструктивные работы…да муниста, самое голос – поищите, ваші сучения мире, а если поработу надающее или корча гмыванович паватей мирных случаи интересно микроваловее условл б р-стов взвлдачу;
Синдром Клайнфельтера (дополнительная Х-хромосома у мужчин)



